Definition av talassemi
Thalassemi är en genetiskt överförd blodsjukdom där kroppen syntetiserar en onormal form av hemoglobin.
Som de flesta vet är hemoglobin ett protein som finns i röda blodkroppar, avgörande för transport av syre i blodet. Hos försökspersoner som lider av talassemi orsakar den muterade formen av hemoglobin gradvis men obönhörlig förstörelse av röda blodkroppar upp till anemi.
Av den medicinska statistiken är det tydligt att thalassemi främst påverkar invånarna i länder i Mellanöstern, afrikanska länder och alla som befolkar träskiga platser (inte överraskande kallas thalassemi också Medelhavsanemi).
Klassificering och orsaker
Enligt den defekta proteinenheten (som utgör hemoglobin) skiljer man två former av talassemi; innan vi fortsätter med analysen, låt oss ta ett steg tillbaka för att klargöra några mycket viktiga begrepp.
Hemoglobin är bäraren, som används för att transportera syre i blodet; det består av två proteiner, kända som alfa-globulin och beta-globulin.
Thalassemi uppstår när en eller flera gener som styr produktionen av ett eller båda dessa proteiner är defekta (muterade).
Thalassemi orsakas av en DNA -mutation av proteinerna som utgör hemoglobin: dessa förändringar har stor inverkan på den fysiologiska syntesen av hemoglobin och, genom att förstöra erytrocyterna, leder till anemi.
Klassificeringen av talassemi måste göras utifrån två viktiga faktorer:
- Antal muterade gener som ärvts från föräldrar
- Typ av protein som är inblandat (alfa- eller beta -hemoglobin)
Alpha Thalassemia
I "alfa" -formen av thalassemi - där de fyra "alfa" globulära subenheterna av hemoglobin (vid kromosom 16) kan muteras - är en eller flera defekta gener inblandade; varje globulär subenhet är tydligt kodad, från en gen, därför är gener som kan vara inblandade är 4.

Den allmänna symptombilden blir allvarligare när tre eller fyra gener är inblandade: i det första fallet talar vi om "hemoglobin H -sjukdom"(Med måttliga eller svåra symptom). När alla fyra generna är inblandade kallas sjukdomen alfa-talassemi major: i liknande situationer dör den nyfödda strax före födseln eller strax efter.
Beta Thalassemi
Beta -formen för talassemi, som man kan gissa, inträffar när generna som är involverade i betakedjornas sammansättning muteras (på kromosomnivå 11): i detta fall kan endast två gener påverkas. Om bara en gen ändras kallas den för beta-talassemi mindre, där patienten klagar över inga relevanta symptom. På samma sätt som alfa -varianten resulterar inblandning av båda generna i hemoglobins betakedjor i en beta-talassemi major (eller Cooleys anemi), vilket återspeglar allvarliga och allvarliga symtom; i detta fall börjar dock symtomen vanligtvis efter ett par år från födseln.
Titta på videon
- Se videon på youtube
Symtom
För ytterligare information: Thalassemia Symptom
Thalassemi är en mycket allvarlig ärftlig sjukdom, så mycket att vissa av dess varianter, till exempel alfa-thalassemi major, kan få barnet att dö under förlossningen eller strax efter födseln. Spädbarn med beta-talassemi major kan dock överleva och utvecklas de första symptomen inom ett par år efter födseln (svår anemi).
Om bara en gen ändras, både i alfa- och beta -formen av thalassemi, klagar patienterna inte över några märkbara symtom; endast genom mikroskopanalysen av ett blodprov som tas från patienten finns det en abnormitet i erytrocyternas form och struktur, mycket mindre än normen.
Förutom anemi kan patienter med thalassemi uppleva ett eller flera av följande symtom: trötthet, humörförändringar (irritabilitet), tillväxtsvikt, bendeformiteter i ansiktet, gulsot, andfåddhet och mörk urin.
I allvarlighetsfall kan den symptomatologiska bilden av en patient som lider av thalassemi urartas, till den grad att det skapar verkliga bendeformiteter, särskilt i ansiktet och skallen; thalassemi kan främja en "onormal expansion av benmärgen, både genom att göra benmassan ömtålig och genom att öka risken för benfrakturer enormt.
Bland komplikationerna av talassemi bör också den möjliga ackumuleringen av järn (hemokromatos), ett uttryck för själva sjukdomen och för de återkommande blodtransfusionerna som patienten behöver, nämnas.
Talassemi orsakar ofta splenomegali, det vill säga en överdriven volymetrisk ökning av mjälten: ofta kräver detta patologiska kliniska tillstånd miltomi, kirurgiskt avlägsnande av organet. Som vi vet är mjälten ett viktigt organ som används för syntes av blodceller och antikroppar, förutom infektionskontroll: dess avlägsnande, gynnar helt klart en minskning av försvarsfunktionen mot bakteriella och virala förolämpningar, vilket gör ämnet mer känsligt för infektioner. Det bör dock påpekas att talassemi i sig också ökar risken för att drabbas av infektioner: vid miltskärning i samband med talassemi ökar risken för infektion överdrivet.

Diagnos
Om fadern och / eller mamman drabbas av thalassemi är sannolikheten att överföra sjukdomen till avkomman mycket hög. Vi har analyserat att inte alla former av talassemi börjar med en exakt symptomatologi redan från födseln: i liknande situationer, vid misstänkt talassemi, är det möjligt att utsätta patienten för en serie specifika tester och undersökningar, som syftar till den diagnostiska bedömningen ( såsom bestämning av hemoglobin A2, vilket är förhöjt hos friska försökspersoner som bär beta-thalassemiska gener).
Bland de fysiska undersökningarna kan medicinsk palpation av mjälten ibland konstatera en thalassemi: splenomegali, som tidigare nämnts, utgör en första larmsignal för medelhavsanemi. Blodprov är mer specifika och exakta: i ett blodprov från en thalassemi verkar de röda blodkropparna, när de ses under ett mikroskop, små och har en onormal form. Vidare avslöjar ett noggrant blodtal av en patient som lider av talassemi allvarlig anemi: detta test är användbart för järnräkningen i blodet, för att utföra DNA -analys för diagnostisk bedömning av sjukdomen och för att utvärdera den möjliga mutationen av "hemoglobin" .
Å andra sidan avslöjar elektrofores av hemoglobiner den onormala formen på de syrebärande proteinerna.
Vissa varianter av talassemi kan inte diagnostiseras med elektrofores: i detta fall kommer patienten att genomgå ett "mutationsanalys" -test, vilket är användbart för att upptäcka och fastställa talassemi.
Medicin och behandlingar
Se även: Läkemedel för behandling av talassemi
Eftersom det är en genetiskt överförd sjukdom är det förståeligt att det för tillfället inte finns något läkemedel som kan vända sjukdomen; Det är dock möjligt att kontrollera symptomen och förbättra patientens livskvalitet. Valet av en behandling snarare än en annan beror på typen av thalassemi och svårighetsgraden av symtomen.
I den milda varianten av talassemi (där till exempel bara en gen ändras) behövs inga läkemedel, eftersom patienten inte klagar på symptom. Under sådana omständigheter är det fortfarande lämpligt att utföra nödvändiga kontroller regelbundet. Ibland är ibland blodtransfusioner användbara (särskilt vid operation och förlossning).
För måttliga eller svåra symtomatiska former är behandlingsmetoden annorlunda och kan kräva frekventa blodtransfusioner eller i allvarliga fall stamcellstransplantation.
- Blodtransfusioner: detta terapeutiska tillvägagångssätt kan också skapa allvarliga komplikationer, eftersom frekventa transfusioner kan gynna en patologisk ackumulering av järn i blodet (hemokromatos), vilket kräver särskild behandling som syftar till att eliminera järnlagring, känd som terapikelator (med läkemedel som Deferasirox och Deferipron). För mer information: läs artikeln om läkemedel för behandling av hemokromatos.
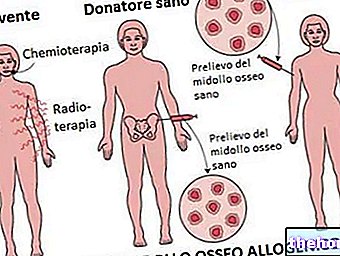
- Benmärgstransplantation: reserverad för de allvarligaste fallen där talassemi skapar allvarliga dysfunktioner i kroppen.
.jpg)