Aktiva ingredienser: Ranibizumab
Lucentis 10 mg / ml injektionsvätska, lösning
Lucentis förpackningsinsatser är tillgängliga för förpackningsstorlekar:- Lucentis 10 mg / ml injektionsvätska, lösning
- Lucentis 10 mg / ml injektionsvätska, lösning i förfylld spruta
Indikationer Varför används Lucentis? Vad är det för?
Vad är Lucentis
Lucentis är en lösning som ska injiceras i ögat. Lucentis är en av en grupp läkemedel som kallas antineovaskulära medel. Den innehåller en aktiv substans som kallas ranibizumab.
Vad är Lucentis för
Lucentis används hos vuxna för att behandla olika ögonsjukdomar som orsakar nedsatt syn.
Dessa tillstånd beror på skador på näthinnan (det ljuskänsliga lagret på ögats baksida) orsakat av:
- Tillväxt av onormala blodkärl som släpper ut vätskor (koroidal neovaskularisering, CNV). Detta ses vid tillstånd som åldersrelaterad makuladegeneration (AMD) eller patologisk närsynthet (PM).
- Makulaödem (svullnad i mitten av näthinnan). Denna svullnad kan orsakas av diabetes (ett tillstånd som kallas diabetiskt makulaödem (DME)) eller av blockering av näthinnorna (ett tillstånd som kallas retinalvenokockulation (RVO)).
Hur Lucentis fungerar
Lucentis känner specifikt igen och binder ett protein som kallas humant endotelkärlstillväxtfaktor A (VEGF-A) som finns i ögat. Vid överskott orsakar VEGF-A onormal blodkärlstillväxt och svullnad i ögat vilket kan leda till minskning av blodkärlen . av syn vid förhållanden som AMD, PM, DME eller RVO. Genom att binda VEGF-A kan Lucentis blockera dess verkan och förhindra onormal tillväxt och svullnad.
Under dessa förhållanden kan Lucentis hjälpa till att stabilisera och i många fall förbättra synen.
Kontraindikationer När Lucentis inte ska användas
Du får inte ta emot Lucentis
- om du är allergisk mot ranibizumab eller något annat innehållsämne i detta läkemedel
- om du har en "infektion i ett öga eller det omgivande området.
- om du har smärta eller rodnad (svår intraokulär inflammation) i ett öga.
Försiktighetsåtgärder vid användning Vad du behöver veta innan du tar Lucentis
Tala med din läkare innan du får Lucentis.
- Lucentis ges som en "injektion i" ögat. Ibland kan en "infektion i den inre delen av ögat", smärta eller rodnad (inflammation) lossna eller brista i ett av lagren i ögats baksida (näthinnelösning eller bristning och lossning eller bristning av epitelet) efter behandling med Lucentis. retinalpigment) eller grumling av linsen (grå starr). Det är viktigt att identifiera och behandla en "näthinnesinfektion eller avlägsnande så snart som möjligt. Tala omedelbart för din läkare om du upplever tecken som ögonsmärta eller ökat obehag, förvärrade röda ögon", suddig eller minskad syn, ökning av kardiovaskulära i syn eller ökad ljuskänslighet.
- Hos vissa patienter kan trycket i ögat öka en kort stund omedelbart efter injektionen. Denna händelse är något du kanske inte märker, så din läkare bör kontrollera efter varje injektion.
- Tala om för din läkare om du tidigare har haft ögonproblem eller behandlingar, eller om du har haft stroke eller tecken på övergående ischemiska attacker (svaghet eller förlamning av lemmar eller ansikte, svårigheter att tala eller förstå). Denna information kommer att beaktas vid utvärderingen av om Lucentis är lämplig behandling för dig.
Barn och ungdomar (under 18 år)
Användningen av Lucentis hos barn och ungdomar har inte studerats och rekommenderas därför inte.
Interaktioner Vilka läkemedel eller livsmedel kan förändra effekten av Lucentis
Tala om för din läkare om du använder, nyligen har använt eller kan tänkas använda andra läkemedel.
Varningar Det är viktigt att veta att:
Graviditet och amning
- Kvinnor i fertil ålder bör rådas att använda effektivt preventivmedel under behandlingen.
- Det finns ingen erfarenhet av användning av Lucentis till gravida kvinnor, därför är de potentiella riskerna inte kända. Om du är gravid, tror att du kan vara gravid eller planerar att bli gravid, diskutera detta med din läkare innan behandling med Lucentis.
- Användning av Lucentis under amning rekommenderas inte eftersom det inte är känt om Lucentis utsöndras i bröstmjölk. Fråga din läkare eller apotekspersonal om råd innan behandling med Lucentis.
Köra och använda maskiner
Tillfällig suddig syn kan uppstå efter behandling med Lucentis. Om detta händer ska du inte köra bil eller använda maskiner förrän detta tillstånd har löst sig.
Dos, metod och administreringstid Hur man använder Lucentis: Dosering
Hur Lucentis kommer att ges till dig
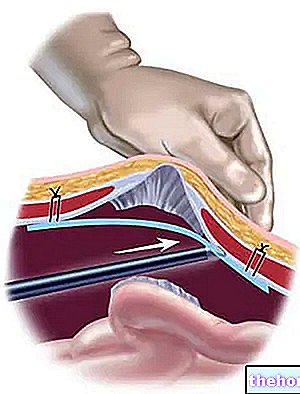
Lucentis ges av din ögonläkare som en enda injektion i ögat under lokalbedövning. Vanlig dos av en injektion är 0,05 ml (som innehåller 0,5 mg av den aktiva ingrediensen). Intervallet mellan två doser som injiceras i samma öga ska vara minst fyra veckor.Alla injektioner ges av din ögonläkare.
Före injektionen kommer din läkare att rengöra ögat noggrant för att förhindra en infektion. Din läkare kommer också att ge dig en lokalbedövning för att minska eller förhindra smärta som kan uppstå vid injektionen.
Behandlingen började med en injektion av Lucentis per månad. Läkaren kommer att övervaka ögats tillstånd och, baserat på svaret på behandlingen, avgöra om och när ytterligare behandling behövs.
Detaljerade instruktioner för användaren finns i slutet av denna bipacksedel under "Hur man förbereder och administrerar Lucentis".
Äldre patienter (65 år och äldre)
Lucentis kan användas för patienter 65 år och äldre utan dosjusteringar.
Om du saknar en dos Lucentis
Kontakta din läkare eller sjukhus så snart som möjligt för att boka om din tid.
Innan behandlingen med Lucentis avbryts
Om du funderar på att sluta behandlingen med Lucentis, gå till ditt nästa besök och diskutera detta med din läkare. Din läkare kommer att ge dig råd och bestämma hur länge du kommer att behöva behandlas med Lucentis.
Fråga din läkare om du har ytterligare frågor om användningen av detta läkemedel.
Biverkningar Vilka är biverkningarna av Lucentis
Liksom alla läkemedel kan detta läkemedel orsaka biverkningar men alla användare behöver inte få dem.
Biverkningar som är förknippade med administrering av Lucentis beror på både själva medicinen och injektionsproceduren och påverkar mest ögat.
De allvarligaste biverkningarna beskrivs nedan:
Vanliga allvarliga biverkningar (kan förekomma hos upp till 1 av 10 patienter): lösgörande eller rivning i ögats baksida (näthinneavlossning eller bristning), manifesterad av ljusglimtar med flottörer upp till en tillfällig synförminskning eller grumling av lins (grå starr.) Mindre vanliga allvarliga biverkningar (kan drabba upp till 1 av 100 patienter): blindhet, infektion i ögongloben (endoftalmitis) med inflammation inuti ögat.
Symptomen du kan uppleva beskrivs i avsnitt 2 i denna bipacksedel (läs avsnitt 2 "Vad du behöver veta innan du får Lucentis"). Kontakta din läkare omedelbart om någon av dessa biverkningar uppstår.
De vanligaste biverkningarna beskrivs nedan:
Mycket vanliga biverkningar (kan förekomma hos fler än 1 av 10 patienter)
Visuella biverkningar inkluderar: inflammation i ögat, blödning på baksidan av ögat (näthinneblödning), synstörningar, ögonsmärta, partiklar eller synfläckar (floaters), lokal ögonrödhet, ögonirritation, kroppskänsla främmande ämnen i öga, ökad tårproduktion, inflammation eller infektion i ögonlockets marginal, torra ögon, röda eller kliande ögon och ökat tryck i ögat.
Icke-visuella biverkningar inkluderar: ont i halsen, nästäppa, rinnande näsa, huvudvärk och ledvärk.
Andra biverkningar som kan uppstå efter behandling med Lucentis beskrivs nedan:
Vanliga biverkningar
Visuella biverkningar inkluderar: minskad synskärpa, svullnad av en del av ögat (uvea, hornhinna), inflammation i hornhinnan (ögats framsida), små märken på ögats yta, dimsyn, blödning vid injektionen, blödning i ögat, kliande urladdning från ögat, rodnad och svullnad (konjunktivit), ljuskänslighet, ögonbesvär, svullnad i ögonlocken, smärta i ögonlocket. Icke-visuella biverkningar inkluderar: urinvägsinfektion, minskade röda blodkroppar (med symtom som t.ex. trötthet, andfåddhet, yrsel, blekhet), ångest, hosta, illamående, allergiska reaktioner som utslag, nässelfeber, klåda och rodnad i huden.
Mindre vanliga biverkningar
Visuella biverkningar inkluderar: inflammation och blödning i ögats framsida, ansamling av pus i ögat, förändringar i den centrala delen av ögonytan, smärta eller irritation vid injektionsstället, onormal känsla i ögat, ögonlockirritation.
Tala med din läkare om du får några biverkningar. Detta inkluderar eventuella biverkningar som inte nämns i denna bipacksedel.
Rapportering av biverkningar
Tala med din läkare om du får några biverkningar. Detta inkluderar eventuella biverkningar som inte nämns i denna bipacksedel. Du kan också rapportera biverkningar direkt via det nationella rapporteringssystemet som anges i bilaga V. Genom att rapportera biverkningar kan du hjälpa till att ge mer information om säkerheten för detta läkemedel.
Giltighetstid och lagring
- Förvara detta läkemedel utom syn- och räckhåll för barn.
- Använd inte detta läkemedel efter utgångsdatum som anges på kartongen och injektionsflaskans etikett efter EXP och efter EXP. Utgångsdatumet avser den sista dagen i den månaden.
- Förvara i kylskåp (2? C - 8? C). Frys inte.
- Förvara injektionsflaskan i ytterkartongen för att skydda läkemedlet från ljus.
- Använd inte förpackningar som är skadade.
Förpackningens innehåll och annan information
Vad Lucentis innehåller
- Den aktiva substansen är ranibizumab (10 mg / ml). Varje ml innehåller 10 mg ranibizumab.
- Övriga ingredienser är a, a-trehalosdihydrat; histidinhydroklorid, monohydrat; histidin; polysorbat 20; vatten för injektionsvätskor.
Beskrivning av hur Lucentis ser ut och förpackningens innehåll
Lucentis är en lösning för injektion i en injektionsflaska (0,23 ml). Lösningen är vattenhaltig, klar, färglös till svagt gul.
Lucentis levereras i en förpackning som innehåller en injektionsflaska med ranibizumab med en klorobutylgummipropp, en trubbig filternål (18G x 1½ ″, 1,2 mm x 40 mm, 5 mikrometer) för uttag av flaskans innehåll, en injektionsnål (30G x ½ ″, 0,3 mm x 13 mm) och en spruta (1 ml) för att ta ut innehållet i injektionsflaskan för intravitreal injektion. Alla komponenter är endast för engångsbruk.
Följande information är endast avsedd för vårdpersonal: Se även avsnittet "Hur Lucentis kommer att ges till dig".
Hur man förbereder och administrerar Lucentis
Injektionsflaskor för engångsbruk endast för intravitreal användning ska administreras av en kvalificerad ögonläkare med erfarenhet av intravitreala injektioner.
Vid våt AMD, synnedsättning på grund av DME, makulaödem sekundärt till RVO eller CNV sekundärt till PM, är den rekommenderade dosen Lucentis 0,5 mg i en enda intravitreal injektion.
Detta motsvarar en injicerad volym på 0,05 ml. Intervallet mellan två doser injicerade i samma öga ska vara minst fyra veckor.
Behandlingen startas med en injektion per månad tills maximal synskärpa uppnås och / eller det inte finns några tecken på sjukdomsaktivitet som förändringar i synskärpa och förändringar av andra tecken och symtom på sjukdomen under fortsatt behandling. Patienter med våt AMD, DME och RVO, kan det vara nödvändigt att påbörja behandling med tre eller flera på varandra följande månatliga injektioner.
Därför bör övervaknings- och behandlingsintervall bestämmas av läkaren och bör baseras på sjukdomsaktivitet, vilket fastställs genom bedömning av synskärpa och / eller anatomiska parametrar.
Om synskärpa och anatomiska parametrar enligt läkarens uppfattning indikerar att patienten inte gynnas av fortsatt behandling, ska Lucentis avbrytas.
Övervakning av sjukdomsaktivitet kan innefatta klinisk undersökning, funktionella bedömningar eller bildtekniker (såsom optisk koherens -tomografi eller fluoresceinangiografi).
Om patienter behandlas enligt en behandlings-och-förlängningsplan, när de når maximal synskärpa och / eller i avsaknad av tecken på sjukdomsaktivitet, kan behandlingsintervallen gradvis förlängas tills tecken på sjukdomen eller det försämras synen fungera. Behandlingsintervallet bör gradvis förlängas med högst två veckor för patienter med våt AMD och kan förlängas upp till en månad för patienter med DME. Behandlingsintervallen kan också gradvis förlängas vid behandling av RVO, men inte där är tillräckliga data för att fastställa varaktigheten av dessa intervaller. När sjukdomsaktiviteten återuppstår bör behandlingsintervallet förkortas i enlighet därmed.
Vid behandling av synskada orsakad av CNV sekundärt till PM kräver många patienter endast en eller två injektioner under det första året, medan vissa kräver mer frekvent behandling.
Lucentis och laserfotokoagulering i DME och makulaödem sekundärt till BRVO
Det finns viss erfarenhet av att Lucentis administreras samtidigt med laserfotokoagulering. Vid användning samma dag ska Lucentis administreras minst 30 minuter efter laserfotokoagulering. Lucentis kan administreras till patienter som tidigare fått laserfotokoagulering.
Lucentis och fotodynamisk terapi med Visudyne i CNV sekundärt till PM
Det finns ingen erfarenhet av administrering av Lucentis i kombination med Visudyne. Före administrering bör Lucentis kontrolleras visuellt med avseende på förekomst av partiklar och missfärgning.
Injektionsproceduren bör utföras under aseptiska förhållanden, inklusive kirurgisk handdesinfektion, sterila handskar, en steril drapering och steril blefarostat (eller motsvarande), och förmågan att utföra en steril paracentes (om det behövs). Utföra det intravitreala förfarandet, patientens medicinsk historia bör utvärderas noggrant för överkänslighetsreaktioner. Tillräcklig anestesi och ett brett spektrum topiskt antimikrobiellt medel bör administreras före injektion för att desinficera den periokulära, okulära och ögonlockiga ytan enligt klinisk praxis.
För att förbereda Lucentis för intravitreal injektion, följ instruktionerna nedan:
Alla komponenter är sterila och endast för engångsbruk. Komponenter med förpackningar som visar tecken på skada eller manipulering får inte användas. Sterilitet kan inte garanteras om komponentförpackningstätningen inte är intakt.
- Desinficera utsidan av injektionsflaskans gummipropp före insamling.
- Fäst aseptiskt den 5 µm filternålen (18G x 1½ ″, 1,2 mm x 40 mm, 5 µm, medföljer) på en 1 ml spruta (medföljer). För in den trubbiga filternålen i mitten av locket tills den berör botten av injektionsflaskan.
- Ta ut all vätska från injektionsflaskan genom att hålla den upprätt, något lutad för att underlätta fullständig uttagning.
- Se till att sprutans kolv dras tillräckligt långt tillbaka när du tömmer injektionsflaskan för att helt tömma filternålen.
- Låt filternålen vara trubbig i injektionsflaskan och ta bort sprutan från den. Kassera filternålen efter att innehållet i injektionsflaskan tagits ut och använd den inte för intravitreal injektion.
- Montera injektionsnålen (30G x ½ ″, 0,3 mm x 13 mm, medföljer) säkert och aseptiskt på sprutan.
- Ta försiktigt av locket från injektionsnålen utan att koppla injektionsnålen från sprutan. Obs! Håll injektionsnålens gula bas medan du tar av locket.
- Driv försiktigt ut luften från sprutan och justera dosen till 0,05 ml markerad på sprutan. Sprutan är klar för injektion. Obs: Rengör inte injektionsnålen. Dra inte kolven bakåt. Sätt in injektionsnålen 3,5-4,0 mm bakom limbus, in i glaskammaren, undvik den horisontella meridianen och rikta nålen mot jordens mitt. Injicera injektionsvolymen på 0,05 ml; ändra skleralplatsen för efterföljande injektioner. Efter injektionen ska du inte täcka nålen eller ta bort den från sprutan. Kassera den använda sprutan tillsammans med nålen i en lämplig behållare eller i enlighet med lokala krav.
Bipacksedel: AIFA (Italian Medicines Agency). Innehåll publicerat i januari 2016. Den information som finns finns kanske inte uppdaterad.
För att få tillgång till den senaste versionen är det lämpligt att gå till AIFA (Italian Medicines Agency) webbplats. Ansvarsfriskrivning och användbar information.
01.0 LÄKEMEDLETS NAMN
LUCENTIS 10 mg / ml injektionsvätska, lösning
02.0 KVALITATIV OCH KVANTITATIV SAMMANSÄTTNING
En ml innehåller 10 mg ranibizumab *. Varje injektionsflaska innehåller 2,3 mg ranibizumab i 0,23 ml lösning.
* Ranibizumab är ett humaniserat monoklonalt antikroppsfragment som produceras i cellerna i Escherichia coli genom rekombinant DNA -teknik.
För fullständig förteckning över hjälpämnen, se avsnitt 6.1.
03.0 LÄKEMEDELSFORM
Injicerbar lösning
Klar, färglös till svagt gul vattenlösning.
04.0 KLINISK INFORMATION
04.1 Terapeutiska indikationer
Lucentis är indicerat för vuxna för:
• Behandling av åldersrelaterad neovaskulär (våt) makuladegeneration (AMD)
• Behandling av synskada orsakad av diabetiskt makulaödem (DME)
• Behandling av synskada orsakad av "makulaödem sekundärt till retinal venocklusion (gren RVO eller central RVO)
• Behandling av synskada orsakad av koroidal neovaskularisering (CNV) sekundärt till patologisk närsynthet (PM)
04.2 Dosering och administreringssätt
Lucentis ska administreras av en kvalificerad ögonläkare med erfarenhet av intravitreala injektioner.
Dosering för behandling av våt AMD
Den rekommenderade dosen Lucentis är 0,5 mg administreras varje månad som en enda intravitreal injektion. Detta motsvarar en injicerad volym på 0,05 ml.
Behandlingen administreras varje månad och fortsätter tills maximal synskärpa har uppnåtts, dvs patientens synskärpa är stabil under tre på varandra följande månatliga kontroller som utförts under ranibizumab -behandling.
Därför bör patienternas synskärpa övervakas varje månad.
Behandlingen ska återupptas när övervakning indikerar en minskning av synskärpan på grund av våt AMD. Månadsinjektioner bör sedan administreras tills stabil synskärpa uppnås igen för tre på varandra följande månatliga kontroller (detta innebär minst två injektioner). Intervallet mellan två doser bör inte vara mindre än en månad.
Dosering för behandling av synskada orsakad av DME eller makulaödem sekundärt till RVO
Den rekommenderade dosen Lucentis är 0,5 mg administreras varje månad som en enda intravitreal injektion. Detta motsvarar en injicerad volym på 0,05 ml.
Behandlingen administreras varje månad och fortsätter tills maximal synskärpa har uppnåtts, dvs patientens synskärpa är stabil under tre på varandra följande månatliga kontroller som utförts under ranibizumab -behandling. Om det inte sker någon förbättring av synskärpa under perioden med de tre första injektionerna, rekommenderas inte fortsatt behandling.
Därför bör patienternas synskärpa övervakas varje månad.
Behandlingen ska återupptas när övervakning indikerar minskad synskärpa på grund av DME eller makulaödem sekundärt till RVO. Månadsinjektioner bör sedan administreras tills stabil synskärpa uppnås igen för tre månadskontroller. I följd (detta innebär minst två injektioner). Intervallet mellan de två doserna bör inte vara mindre än en månad.
Lucentis och laserfotokoagulering i DME och makulaödem sekundärt till BRVO
Det finns viss erfarenhet av administrering av Lucentis samtidigt med laserfotokoagulering (se avsnitt 5.1). När det ges samma dag ska Lucentis ges minst 30 minuter efter laserfotokoagulering. Lucentis kan ges till patienter som tidigare fått laserfotokoagulering.
Dosering för behandling av synskada orsakad av CNV sekundärt till PM
Behandlingen bör startas med en enda injektion.
Om övervakning avslöjar tecken på sjukdomsaktivitet, såsom nedsatt synskärpa och / eller tecken på skada, rekommenderas ytterligare behandling.
Sjukdomsövervakning kan innefatta en klinisk undersökning, optisk koherens tomografi (OCT) eller fluorescein angiografi (FA).
Vissa patienter kanske bara behöver en eller två injektioner under det första behandlingsåret, men vissa kan behöva mer frekvent behandling (se avsnitt 5.1). Månadsövervakning rekommenderas därför under de första två månaderna och minst var tredje månad under det första behandlingsåret. Efter det första året kan övervakningsfrekvensen bestämmas av läkaren.
Intervallet mellan två doser bör inte vara mindre än en månad.
Lucentis och fotodynamisk terapi med Visudyne i CNV sekundärt till PM
Det finns ingen erfarenhet av administrering av Lucentis i kombination med Visudyne.
Särskilda populationer
Leverinsufficiens
Lucentis har inte studerats hos patienter med leverinsufficiens. Inga särskilda överväganden behövs dock för denna polering.
Njursvikt
Ingen dosjustering är nödvändig hos patienter med njurinsufficiens (se avsnitt 5.2).
Pensionärer
Ingen dosjustering krävs hos äldre. Det finns "begränsad" erfarenhet av patienter med DME över 75 år.
Pediatrisk population
Säkerhet och effekt för Lucentis hos barn och ungdomar under 18 år har inte fastställts. Det finns inga tillgängliga data.
Administreringssätt
Injektionsflaskor för engångsbruk endast för intravitreal användning.
Före administrering bör Lucentis kontrolleras visuellt med avseende på förekomst av partiklar och missfärgning.
Injektionsproceduren måste utföras under aseptiska förhållanden, som inkluderar desinfektion av händerna som för alla kirurgiska ingrepp, sterila handskar, en steril draperi och en steril blefarostat (eller motsvarande) och möjligheten att utföra en steril paracentes (om det behövs patientens historia av överkänslighetsreaktioner bör utvärderas noggrant före det intravitreala förfarandet (se avsnitt 4.4). Tillräcklig anestesi och ett brett spektrum topiskt antimikrobiellt medel bör administreras före injektion för att desinficera den periokulära, okulära och ögonlockiga ytan enligt klinisk praxis.
För information om beredning av Lucentis, se avsnitt 6.6.
Sätt in injektionsnålen 3,5-4,0 mm bakom limbus, i glaskammaren, undvik den horisontella meridianen och rikta nålen mot mitten av ögongloben. Injicera injektionsvolymen på 0,05 ml; byt skleralt ställe för efterföljande injektioner.
04.3 Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt 6.1.
Patienter med nuvarande eller misstänkta okulära eller periokulära infektioner.
Patienter med pågående svår intraokulär inflammation.
04.4 Särskilda varningar och lämpliga försiktighetsåtgärder vid användning
Reaktioner relaterade till intravitreal injektion
Intravitreala injektioner, inklusive de med Lucentis, har associerats med endoftalmitis, intraokulär inflammation, rhegmatogen retinalavlossning, retinalruptur och iatrogen traumatisk grå starr (se avsnitt 4.8). Lämpliga aseptiska injektionstekniker bör alltid användas för administrering av Lucentis. Dessutom ska patienter övervakas i veckan efter injektionen för att möjliggöra snabb behandling vid infektion. Patienter bör instrueras om hur de ska rapportera alla symptom som tyder på endoftalmitis eller någon av ovanstående händelser utan dröjsmål.
Ökning av intraokulärt tryck
Övergående ökningar av intraokulärt tryck (IOP) har observerats inom 60 minuter efter Lucentis -injektion. Långvariga ökningar av IOP har också observerats (se avsnitt 4.8). Intraokulärt tryck och perfusion av synnerven skall övervakas och behandlas på lämpligt sätt.
Bilateral behandling
Begränsade data om bilateral användning av Lucentis (inklusive dosering samma dag) tyder inte på en ökad risk för systemiska biverkningar jämfört med ensidig behandling.
Immunogenicitet
Det finns en potential för immunogenicitet med Lucentis. Eftersom det finns en möjlighet till ökad systemisk exponering hos personer med DME, kan en ökad risk för att utveckla överkänslighet hos denna patientpopulation inte uteslutas.Patienter bör också utbildas i hur man rapporterar om intraokulär inflammation förvärras eftersom det kan vara ett kliniskt symptom som kan hänföras till bildandet av intraokulära antikroppar.
Samtidig användning med andra anti-VEGF (vaskulär endotel tillväxtfaktor)
Lucentis får inte administreras samtidigt med andra anti-VEGF-läkemedel (systemiska eller okulära).
Lucentis avbryter
Dosen ska inte administreras och behandlingen ska inte återupptas före nästa schemalagda behandling vid:
• en minskning av bäst korrigerad synskärpa (BCVA) ≥30 bokstäver jämfört med den senaste utvärderingen;
• ett intraokulärt tryck ≥30 mmHg;
• en näthinnepaus;
• en "subretinal blödning som sträcker sig till mitten av fovea, eller om blödningens omfattning är ≥50% av det totala lesionsområdet";
• intraokulär kirurgi utförd eller planerad under de föregående eller kommande 28 dagarna.
Bristning av retinalpigmentepitelet
Riskfaktorer förknippade med uppkomsten av retinalpigmentepitelbrott efter anti-VEGF-behandling för våt AMD inkluderar stor och / eller hög retinal pigmentepitelavskiljning.Vid behandling med Lucentis ska försiktighet iakttas hos patienter med dessa riskfaktorer för bristning av retinalpigmentepitelet.
Regmatogen näthinneavlossning eller makulära hål
Behandlingen ska avbrytas hos personer med rhegmatogen retinalavskiljning eller steg 3 eller 4 makulära hål.
Befolkning med begränsad data
Det finns endast begränsad erfarenhet av behandling av patienter med DME som är sekundär till diabetes typ I. Lucentis har inte studerats hos patienter som tidigare fått intravitreala injektioner, hos patienter med aktiva systemiska infektioner, proliferativ diabetisk retinopati eller hos patienter med samtidiga medicinska tillstånd Det finns heller ingen erfarenhet av behandling med Lucentis hos diabetespatienter med HbAlc större än 12% och okontrollerad hypertoni. Brist på information bör övervägas av läkaren vid behandling av dessa patienter.
Hos patienter med PM finns begränsade data om effekten av Lucentis hos patienter som tidigare behandlats med misslyckad fotodynamisk behandling med verteporfin (vPDT) .Vidare, medan en konsekvent effekt observerades hos personer med subfoveala och juxtafoveala lesioner, finns det otillräckliga data om effekt av Lucentis hos PM -patienter med extrafoveala lesioner.
Systemiska effekter efter intravitreal administrering
Systemiska biverkningar inklusive icke-okulära blödningar och arteriella tromboemboliska händelser har rapporterats efter intravitreal injektion av VEGF-hämmare.
Det finns begränsade data om säkerheten vid behandling av DME, makulaödem orsakat av RVO och CNV sekundärt till PM hos patienter med stroke eller övergående ischemiska attacker. Särskild försiktighet bör iakttas vid behandling av sådana patienter (se avsnitt 4.8).
Tidigare avsnitt av RVO, ischemisk gren och central RVO
Det finns begränsad erfarenhet av behandling av patienter med tidigare episoder av RVO och av patienter med ischemisk gren RVO (BRVO) och central RVO (CRVO). Hos patienter med RVO som uppvisar nedsatt synfunktion med kliniska tecken på irreversibel ischemi, behandling rekommenderas inte.
04.5 Interaktioner med andra läkemedel och andra former av interaktion
Inga konventionella interaktionsstudier har utförts.
För kombinerad användning av fotodynamisk terapi (PDT) med verteporfin och Lucentis vid våt AMD och PM, se avsnitt 5.1.
Se avsnitt 4.2 och 5.1 för kombinerad användning av laserfotokoagulering och Lucentis vid behandling av DME och BRVO.
04.6 Graviditet och amning
Kvinnor i fertil ålder / preventivmedel hos kvinnor
Kvinnor i fertil ålder bör använda effektivt preventivmedel under behandlingen.
Graviditet
För ranibizumab finns inga kliniska data om exponerade graviditeter tillgängliga. Studier på cynomolgus apor har inte visat några direkta eller indirekta skadliga effekter med avseende på graviditet eller embryonal / fosterutveckling (se avsnitt 5.3). Systemisk exponering för ranibizumab är låg efter okulär administrering, men på grund av verkningsmekanismen bör ranibizumab anses vara potentiellt teratogent och embryo / foetotoxiskt. Därför ska ranibizumab inte användas under graviditet om inte de förväntade fördelarna uppväger den potentiella risken för fostret. Kvinnor som planerar att bli gravida och har behandlats med ranibizumab rekommenderas att vänta minst 3 månader efter deras sista dos ranibizumab innan de får barn.
Graviditet
Det är inte känt om Lucentis utsöndras i bröstmjölk. Det rekommenderas att inte amma när du använder Lucentis.
Fertilitet
Det finns inga tillgängliga data om fertilitet.
04.7 Effekter på förmågan att framföra fordon och använda maskiner
Lucentis -behandlingsproceduren kan orsaka övergående synstörningar som kan påverka förmågan att framföra fordon eller använda maskiner (se avsnitt 4.8). Patienter som upplever dessa symtom ska inte köra bil eller använda maskiner förrän dessa övergående synstörningar upphör.
04.8 Biverkningar
Sammanfattning av säkerhetsprofilen
De flesta biverkningar som rapporterats efter administrering av Lucentis är relaterade till det intravitreala injektionsförfarandet.
De vanligaste rapporterade okulära biverkningarna efter Lucentis -injektion är: ögonsmärta, okulär hyperemi, ökat intraokulärt tryck, vitreit, glaskroppsavlossning, näthinneblödning, synstörning, floaters (glasögonflödare), konjunktivalblödning, irritation i ögat, främmande kroppskänsla i öga, ökad rivning, blefarit, torra ögon och kliande ögon.
De vanligaste icke-okulära biverkningarna är huvudvärk, nasofaryngit och artralgi.
Mindre vanligt rapporterade men allvarligare biverkningar inkluderar endoftalmitis, blindhet, näthinnelösning, näthinnesbrott och iatrogen traumatisk grå starr (se avsnitt 4.4).
Patienterna bör informeras om symtomen på dessa potentiella biverkningar och instrueras att informera sin läkare om de upplever tecken som ögonsmärta eller ökat obehag, försämring av ögonrödhet, suddig eller minskad syn, ett ökat antal glasögonflytare eller en " ökad ljuskänslighet.
Biverkningar som rapporterats efter administrering av Lucentis i kliniska studier sammanfattas i tabellen nedan.
Biverkningstabell #
Biverkningar listas efter systemorganklass och frekvens enligt följande konvention: mycket vanliga (≥1 / 10), vanliga (≥1 / 100,
Infektioner och angrepp
Väldigt vanligt Nasofaryngit
allmänning Urinvägsinfektion *
Störningar i blodet och lymfsystemet
allmänning Anemi
Störningar i immunsystemet
allmänning Överkänslighet
Psykiatriska störningar
allmänning Ångest
Nervsystemet
Väldigt vanligt Huvudvärk
Ögonbesvär
Väldigt vanligt Vitrit, glaskroppsavskiljning, näthinneblödning, synstörningar, ögonsmärta, glasögonflödare, konjunktivalblödning, ögonirritation, främmande kroppsförnimmelse i ögat, ökad tårbildning, blefarit, torra ögon, okulär hyperemi, kliande ögon.
allmänning Retinal degeneration, näthinnestörningar, näthinneavlossning, näthinneavbrott, lossning av retinalpigmentepitelet, rivning av retinalpigmentepitelet, nedsatt synskärpa, glasögonblödning, glasögonstörningar, uveit, irit, iridocyklit, grå starr, subkapsulär katarakt posterior kapsel, punktering keratit, hornhinneskada, främre kammarreaktion, dimsyn, blödning vid injektionsstället, blödning i ögonen, konjunktivit, konjunktivit
allergisk, okulär urladdning, ljusblixtar, fotofobi, obehag i ögat, ögonlock i ögonlocket, smärta i ögonlocken, konjunktival hyperemi.
Ovanlig Blindhet, endoftalmitis, hypopion, hyphema, keratopati, iris synechiae, hornhinneavlag, hornhinnödem, hornhinnestriar, smärta vid injektionsstället, irritation på injektionsstället, onormal känsla i ögat, ögonlockirritation.
Andningsvägar, bröstkorg och mediastinum
allmänning Hosta
Gastrointestinala störningar
allmänning Illamående
Hud och subkutan vävnad
allmänning Allergiska reaktioner (utslag, nässelfeber, klåda, erytem)
Muskuloskeletala systemet och bindvävssjukdomar
Väldigt vanligt Artralgi
Diagnostiska tester
Väldigt vanligt Ökat intraokulärt tryck
# Biverkningar definierades som biverkningar (hos minst 0,5 procentenheter av patienter) som inträffade i högre takt (minst 2 procentenheter) hos patienter som fick behandling med Lucentis 0,5 mg jämfört med dem som fick kontrollbehandling (sken eller PDT verteporfin).
* observeras endast i populationen med DME
Biverkningar relaterade till läkemedelskategori
I fas III-våta AMD-studier ökade den totala frekvensen av icke-okulära blödningar, en biverkning som potentiellt kan relateras till VEGF-hämmare (endotelkärlstillväxtfaktor), något hos patienter som behandlats med ranibizumab, men det finns ingen. mönster mellan de olika blödningarna. Det finns en teoretisk risk för arteriella tromboemboliska händelser, inklusive stroke och hjärtinfarkt, till följd av intravitreal användning av VEGF -hämmare. En låg förekomst av arteriella tromboemboliska händelser observerades i kliniska prövningar med Lucentis hos patienter med AMD, DME, RVO och PM och inga skillnader observerades mellan ranibizumabgrupperna jämfört med kontroll.
Rapportering av misstänkta biverkningar
Rapportering av misstänkta biverkningar som inträffar efter godkännande av läkemedlet är viktigt eftersom det möjliggör kontinuerlig övervakning av nytta / riskförhållandet för läkemedlet.Hälso- och sjukvårdspersonal uppmanas att rapportera alla misstänkta biverkningar via den italienska läkemedelsmyndigheten. , webbplats: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Överdosering
Fall av oavsiktlig överdos har rapporterats från kliniska prövningar med våt AMD och data efter marknadsföring. De biverkningar som oftast är förknippade med dessa fall var ökat intraokulärt tryck, övergående blindhet, minskad synskärpa, hornhinnödem och smärta. Om en överdosering inträffar, intraokulärt tryck bör övervakas och behandlas så som läkaren anser nödvändigt.
05.0 FARMAKOLOGISKA EGENSKAPER
05.1 Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Oftalmologiska, anti-neovaskulära medel, ATC-kod: S01LA04
Ranibizumab är ett humaniserat rekombinant monoklonalt antikroppsfragment riktat mot human vaskulär endotel tillväxtfaktor A (VEGF-A). Den binder med hög affinitet till VEGF-A-isoformer (t.ex. VEGF110, VEGF121 och VEGF165), vilket förhindrar bindning av VEGF-A till dess VEGFR-1- och VEGFR-2-receptorer. Till dess receptorer leder till en proliferation av endotelceller till en neovaskularisering, och till en ökning av vaskulär permeabilitet, som antas bidra till utvecklingen av den neovaskulära formen av åldersrelaterad makuladegeneration, patologisk närsynthet eller nedsatt syn orsakad av antingen diabetiskt makulaödem eller "Makulärt ödem sekundärt till RVO.
Behandling av våt AMD
För våt AMD utvärderades säkerheten och den kliniska effekten av Lucentis i tre 24-månaders randomiserade, dubbelblinda, sken- eller aktivkontrollerade studier på patienter med neovaskulär AMD. Totalt 1323 patienter (879 behandlade och 444 kontroller) registrerades i dessa studier.
I studie FVF2598g (MARINA) fick 716 patienter med minimalt klassiska eller ockulta koroidala neovaskulariseringsskador (CNV) utan klassisk komponent månatliga intravitreala injektioner av Lucentis 0,3 mg (n = 238) eller 0,5 mg (n = 240) eller skeninjektioner (n = 238).
I studie FVF2587g (ANCHOR) fick 423 patienter med övervägande klassisk CNV en av följande behandlingar: 1) månatliga intravitreala injektioner av Lucentis 0,3 mg och PDT sham (n = 140); 2) månatliga intravitreala injektioner av Lucentis 0,5 mg och PDT sham (n = 140); eller 3) intravitreala skeninjektioner och PDT med verteporfin (n = 143). PDT med verteporfin eller sken administrerades tillsammans med den initiala injektionen av Lucentis och därefter var tredje månad om fluorangiografi visade uthållighet eller återupptagande av kärlläckage.
De viktigaste resultaten sammanfattas i tabellerna 1, 2 och figur 1.
Tabell 1 Resultat vid månad 12 och månad 24 i studie FVF2598g (MARINA)
ap
Tabell 2 Resultat vid månad 12 och månad 24 i studie FVF2587g (ANKARE)
Tabell
Resultaten av båda studierna visade att fortsatt behandling med ranibizumab också kan vara till nytta för patienter som tappade ≥15 bokstäver med bäst korrigerad synskärpa (BCVA) under det första behandlingsåret.
Studie FVF3192g (PIER) var en randomiserad, dubbelblind, skenkontrollerad studie avsedd att utvärdera säkerheten och effekten av Lucentis hos 184 patienter med alla former av neovaskulär AMD. Patienterna fick intravitreala injektioner av Lucentis 0,3 mg (n = 60) eller 0,5 mg (n = 61) eller skeninjektioner (n = 63) en gång i månaden i 3 doser i följd, följt av en dos som ges var tredje månad. Från månad 14 i studien fick patienter som behandlats med en skeninjektion behandling med ranibizumab och från månad 19 kunde mer frekventa behandlingar utföras. Patienter som behandlades med Lucentis i PIER -studien fick i genomsnitt 10 behandlingar totalt.
Det primära effektmåttet var den genomsnittliga förändringen i synskärpa vid 12 månader jämfört med baslinjen. Efter en initial ökning av synskärpa (efter månadsdos) minskade i genomsnitt patienternas synskärpa med kvartalsdosering och återvände till baslinjen vid månad 12 och denna effekt bibehölls hos majoriteten av de behandlade patienterna. Med ranibizumab (82%) vid Månad 24. Data från ett begränsat antal försökspersoner som hade överförts till ranibizumab -behandling efter mer än ett års skenbehandling tyder på att tidigt behandlingsstart kan vara associerat med bättre bibehållande av "synskärpa.
I både MARINA- och ANCHOR-studierna följde förbättringen av synskärpa som observerades med Lucentis 0,5 mg efter 12 månader av patientrapporterade fördelar mätt av National Eye Institute Visual Function Questionnaire (VFQ-25) poäng. Skillnader mellan Lucentis 0,5 mg och de två kontrollgrupperna utvärderades med p -värden från 0,009 till
Lucentis effekt vid behandling av våt AMD bekräftades i AMD-studier efter marknadsföring. Data från två studier (MONT BLANC, BPD952A2308 och DENALI, BPD952A2309) visade inte ytterligare effekter. Av den kombinerade administreringen av verteporfin (Visudyne PDT) och Lucentis jämfördes till Lucentis ensam.
Behandling av synskada på grund av DME
Säkerhet och effekt för Lucentis utvärderades i två randomiserade, dubbelblinda, skenkontrollerade eller aktiva 12 månaders studier på patienter med nedsatt syn på grund av diabetiskt makulaödem. Totalt av dessa studier registrerades. Av 496 patienter (336 aktiva och 160 kontroller), de flesta hade diabetes typ II, 28 behandlade patienter hade typ I -diabetes.
I fas II i studie D2201 (RESOLVE) behandlades 151 patienter med ranibizumab (6 mg / ml, n = 51, 10 mg / ml, n = 51) eller sken (n = 49) med en "intravitreal injektion per månad. tills fördefinierade kriterier var uppfyllda. Startdosen av ranibizumab (0,3 mg eller 0,5 mg) kunde fördubblas när som helst under studien efter den första injektionen. Laserfotokoagulering tilläts som räddningsbehandling från månad 3 i båda behandlingsarmarna. Studien hade två delar: en undersökande del (de första 42 patienterna besökte vid månad 6) och en bekräftande del (de återstående 109 patienterna besökte vid månad 12).
Viktiga resultat från den bekräftande delen av studien (2/3 av patienterna) sammanfattas i tabell 3.
Tabell 3 Resultat vid månad 12 i studie D2201 (RESOLVE) (total studiepopulation)
ap
I fas III -studie D2301 (RESTORE) randomiserades 345 patienter med nedsatt syn på grund av makulaödem för att antingen få en "intravitreal injektion av 0,5 mg ranibizumab som monoterapi och laser sham -fotokoagulering (n = 116), eller en kombination av 0,5 mg ranibizumab och laserfotokoagulering (n = 118) eller en skeninjektion och laserfotokoagulering (n = 111). Behandlingen med ranibizumab inleddes med månatliga intravitreala injektioner och fortsatte tills synskärpan förblev stabil under minst tre på varandra följande månatliga kontroller. Behandlingen återupptogs när en minskning av BCVA på grund av DME -progression observerades. Laserfotokoagulering administrerades vid baslinjen samma dag, minst 30 minuter före ranibizumab -injektion, och därefter efter behov baserat på ETDRS -kriterier.
De viktigaste resultaten sammanfattas i tabell 4 och figur 2.
Tabell 4 Resultat vid månad 12 i studie D2301 (RESTORE)
ap
Effekten var konsekvent i de flesta undergrupper, men patienter med en ganska hög BCVA vid baslinjen (> 73 bokstäver) med makulaödem och central retinaltjocklek
Förbättringen av synskärpa vid månad 12 observerad med Lucentis 0,5 mg åtföljdes av patientrapporterade fördelar med stora synrelaterade funktioner mätt med National Eye Institute Visual Function Questionnaire (VFQ-25) poäng. Inga skillnader på grund av behandling kan vara fastställd i underklasserna till detta frågeformulär.Skillnaden mellan Lucentis 0,5 mg och kontrollgruppen utvärderades med ett p-värde på 0,0137 (ranibizumab mono) och 0,0041 (ranibizumab + laser) för VFQ-25-kompositpoängen.
I båda studierna åtföljdes den visuella förbättringen av en kontinuerlig minskning av makulaödem mätt som central retinaltjocklek (CRT).
Behandling av synskada orsakad av makulaödem sekundärt till RVO
Den kliniska säkerheten och effekten av Lucentis hos patienter med nedsatt syn på grund av makulaödem sekundärt till RVO utvärderades i randomiserade, dubbelblinda, kontrollerade studier: BRAVO och CRUISE som rekryterade patienter med BRVO (n = 397) och CRVO (n = 392 I båda studierna fick patienterna antingen 0,3 mg eller 0,5 mg ranibizumab intravitreala eller skeninjektioner. Efter 6 månader flyttades patienter i skenkontrollarmen till ranibizumabgruppen 0,5 mg. I BRAVO -studien laserfotokoagulering som räddningsbehandling var tillåtet i alla armar från månad 3.
Nyckelfynd från BRAVO- och CRUISE -studierna presenteras i tabellerna 5 och 6
Tabell 5 Resultat vid månad 6 och 12 (BRAVO)
ap
Tabell 6 Resultat vid månad 6 och 12 (CRUISE)
ap
I båda studierna åtföljdes den visuella förbättringen av en kontinuerlig och signifikant minskning av makulaödem mätt med central tjocklek på näthinnan.
Hos BRVO -patienter (BRAVO -studie och HORIZON -studieförlängning): Efter 2 år hade patienter som hade behandlats med skeninjektioner under de första 6 månaderna och sedan gick över till ranibizumab -behandling en ökning i AV (& symp; 15 bokstäver) jämförbar med den av patienter som hade behandlats med ranibizumab sedan studiestart (& symp; 16 bokstäver). Antalet patienter som avslutade 2 år var dock begränsat och endast kvartalsvisa besök var planerade i HORIZON -studien. det finns tillräckligt med bevis för att avsluta med rekommendationer om när ranibizumab -behandling ska påbörjas hos patienter med BRVO.
Hos CRVO -patienter (CRUISE -studie och HORIZON -studieförlängning): Efter 2 år visade patienter som hade behandlats med skeninjektioner under de första 6 månaderna och sedan överfördes till ranibizumab -behandling inga vinster i AV (& symp; 6 bokstäver) jämfört med dem av patienter som hade behandlats med ranibizumab sedan studiestart (& symp; 12 bokstäver).
Förbättringen av synskärpa som observerades vid behandling med ranibizumab vid månaderna 6 och 12 åtföljdes av patientrapporterade fördelar mätt av National Eye Institute Visual Function Questionnaire (NEI VFQ-25) undergrupp för nära och avlägsna aktiviteter Skillnaden mellan Lucentis 0,5 mg och kontrollgruppen låg mellan p -värden mellan 0,02 och 0,0002.
Behandling av synskada på grund av CNV sekundärt till PM
Säkerheten och den kliniska effekten av Lucentis hos patienter med synskada på grund av CNV i PM validerades baserat på 12-månadersdata från den randomiserade, dubbelblinda, kontrollerade studien F2301 (RADIANCE). Denna studie syftade till att utvärdera två olika doseringsregimer av ranibizumab 0,5 mg administrerat genom intravitreal injektion kontra verteporfin PDT (vPDT, Visudyne fotodynamisk terapi). De 277 patienterna randomiserades till en av följande armar:
• Grupp I (ranibizumab 0,5 mg, behandlingsplan bestämd utifrån "stabilitet" -kriterier definierade som ingen förändring i BCVA jämfört med bedömningar från de två föregående månaderna).
• Grupp II (ranibizumab 0,5 mg, behandlingsplan bestämd utifrån "sjukdomsaktivitet" -kriterier definierade som synskada som kan hänföras till intra- eller subretinal vätska eller aktivt läckage orsakat av CNV-lesioner, vilket framgår av OCT och / eller AF).
• Grupp III (patienter behandlade med vPDT - med möjlighet till behandling med ranibizumab från och med månad 3).
Under studiens 12 månader fick patienterna i genomsnitt 4,6 injektioner (intervall 1-11) i grupp I och 3,5 injektioner (intervall 1-12) i grupp II. Bland patienter som tillhör grupp II, vilket återspeglar den rekommenderade doseringen (se avsnitt 4.2), genomgick 50,9% av patienterna behandling med 1 till 2 injektioner, 34,5% 3 till 5 injektioner och 14,7% gav 6 till 12 injektioner under 12-månadersstudien . 62,9% av grupp II -patienterna krävde inga injektioner under de andra 6 månaderna av studien.
Viktiga resultat från RADIANCE sammanfattas i tabell 7 och figur 5.
Tabell 7 Resultat vid månad 3 och 12 (RADIANCE)
ap
b Jämförande kontroll upp till månad 3. Patienter randomiserade för att få vPDT var berättigade till ranibizumabbehandling vid månad 3 (i grupp III fick 38 patienter ranibizumab vid månad 3)
Förbättringen av synen åtföljdes av en minskning av den centrala näthinnans tjocklek.
Jämfört med den vPDT-behandlade gruppen rapporterade patienter i de ranibizumabbehandlade grupperna fördel (p-värde
Pediatrisk population
Säkerhet och effekt för ranibizumab hos barn har ännu inte fastställts.
Europeiska läkemedelsmyndigheten har avstått från skyldigheten att lämna in resultaten av studier med Lucentis i alla undergrupper av den pediatriska populationen för neovaskulär AMD, synskada på grund av DME, synskada på grund av sekundärt makulaödem till RVO och synskada på grund av CNV sekundärt till PM (se avsnitt 4.2 för information om pediatrisk användning).
05.2 Farmakokinetiska egenskaper
Efter månatlig intravitreal administrering av Lucentis till patienter med neovaskulär AMD var serumkoncentrationerna av ranibizumab generellt låga, med toppnivåer (Cmax) generellt under ranibizumabkoncentrationen som krävs för att hämma den biologiska aktiviteten hos VEGF med 50% (11-27 ng / ml, utvärderas i ett test in vitro cellproliferation). Cmax var dosproportional i hela dosintervallet 0,05 till 1,0 mg / öga. Hos ett begränsat antal patienter med DME indikerar de detekterade serumkoncentrationerna att en något högre systemisk exponering inte kan uteslutas än för de som observerats hos patienter med neovaskulär AMD. Seribalansen för ranibizumab hos patienter med RVO var liknande eller något högre än de som observerades hos patienter med neovaskulär AMD.
Baserat på populationsfarmakokinetisk analys och serumclearance för ranibizumab för neovaskulära AMD-patienter som behandlats med 0,5 mg-dosen, är den genomsnittliga halveringstiden för glasögon för ranibizumab cirka 9 dagar. Vid den månatliga intravitreala administreringen av Lucentis 0,5 mg / öga, förväntas serum C för ranibizumab, cirka 1 dag efter dos, i allmänhet ligga mellan 0,79 och 2,90 ng / ml, medan det förväntas att Cmin i allmänhet varierar mellan 0,07 och 0,49 ng / ml. Seribaserade koncentrationer av ranibizumab beräknas vara cirka 90 000 gånger lägre än glasögonkoncentrationer.
Patienter med nedsatt njurfunktion: Inga konventionella studier har utförts för att undersöka Lucentis farmakokinetik hos patienter med nedsatt njurfunktion. I en "farmakokinetisk analys i en population av neovaskulära AMD -patienter hade 68% (136 av 200) av patienterna" njurinsufficiens (46,5% mild [50-80 ml / min], 20% måttlig [30-50 ml / min) min] och 15% svår [systemiskt clearance var något lägre, men detta var inte kliniskt signifikant.
Patienter med leverinsufficiens: Inga konventionella studier har utförts för att undersöka farmakokinetiken för Lucentis hos patienter med leverinsufficiens.
05.3 Prekliniska säkerhetsdata
Bilateral intravitreal administrering av ranibizumab till cynomolgus apor i doser mellan 0,25 mg / öga och 2,0 mg / öga en gång varannan vecka i upp till 26 veckor resulterade i dosberoende okulära effekter.
Intraokulärt inträffade dosberoende ökningar av flare och celler i den främre kammaren, toppade 2 dagar efter injektionen. Allvarligheten av det inflammatoriska svaret minskar i allmänhet med efterföljande injektioner eller under återhämtningsperioden. som också tenderade att vara dosberoende och i allmänhet kvarstod till slutet av behandlingsperioden.I 26-veckorsstudien ökade svårighetsgraden av glasögoninflammation med antalet injektioner. Reversibilitet observerades dock efter återhämtningsperioden. Arten och varaktigheten av den posteriora segmentinflammationen indikerar ett immunförmedlat antikroppssvar, vilket kan vara kliniskt irrelevant. Kataraktbildning har observerats hos vissa djur efter en relativt lång period av intensiv inflammation, vilket tyder på att linsförändringar var sekundära till svår inflammation.En övergående ökning av intraokulärt tryck observerades efter administrering, oavsett dos, efter intravitreala injektioner.
Mikroskopiska okulära förändringar var relaterade till inflammation och indikerade inte degenerativa processer. Inflammatoriska granulomatösa förändringar noterades i optiska skivor i vissa ögon. Dessa posteriora segmentförändringar minskade och i vissa fall försvann under återhämtningsperioden.
Det fanns inga tecken på systemisk toxicitet efter intravitreal administrering. Serum och glasögonantikroppar mot ranibizumab hittades i en delmängd av behandlade djur.
Det finns inga data om cancerframkallande eller mutagenicitet.
Hos gravida apor orsakade intravitreal injektion av ranibizumab som resulterade i en maximal systemisk exponering 0,9-7 gånger den sämsta kliniska exponeringen inte utvecklingstoxicitet eller teratogenicitet, och hade ingen effekt på kroppens vikt eller struktur. Placenta, även om ranibizumab bör övervägas potentiellt teratogent och embryo / foetotoxiskt baserat på dess farmakologiska effekt.
Frånvaron av medierade effekter av ranibizumab på embryo / fosterutveckling är troligtvis relaterat främst till Fab -fragmentets oförmåga att passera moderkakan. Emellertid beskrevs ett fall med höga moderns serumnivåer av ranibizumab och förekomst av ranibizumab i fosterserum, vilket tyder på att anti-ranibizumab-antikroppen fungerade som ett protein (som innehåller FC-regionen) som transporterar ranibizumab och därigenom minskar dess eliminering från moderns serum och tillåter dess överföring till moderkakan. Eftersom testerna på embryo / fosterutveckling har utförts på friska dräktiga djur och vissa sjukdomar (t.ex. diabetes) kan modifiera placentapermeabiliteten till ett Fab -fragment måste studien tolkas med försiktighet.
06.0 LÄKEMEDELSINFORMATION
06.1 Hjälpämnen
a, a-trehalosdihydrat
Histidinhydroklorid, monohydrat
Histidin
Polysorbat 20
Vatten för injektionsvätskor
06.2 Oförenlighet
I avsaknad av kompatibilitetsstudier får detta läkemedel inte blandas med andra läkemedel.
06.3 Giltighetstid
3 år
06.4 Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 ° C - 8 ° C).
Frys inte.
Förvara injektionsflaskan i ytterkartongen för att skydda läkemedlet från ljus.
06.5 Förpackningens innehåll och förpackningens innehåll
0,23 ml steril lösning i en injektionsflaska (typ I -glas) med propp (klorbutylgummi), 1 trubbig filternål (18G x 1½ ", 1,2 mm x 40 mm, 5 mcm), 1 injektionsnål (30G x ½", 0,3 mm x 13 mm) och 1 spruta (polypropen) (1 ml). Förpackningen innehåller 1 injektionsflaska.
06.6 Anvisningar för användning och hantering
Injektionsflaskan, injektionsnålen, filternålen och sprutan är endast för engångsbruk. Återanvändning kan orsaka infektion eller annan sjukdom / skada. Alla komponenter är sterila. Komponenter med förpackningar som visar tecken på skada eller manipulering får inte användas. Sterilitet kan inte garanteras om komponentförpackningstätningen inte är intakt.
För att förbereda Lucentis för intravitreal injektion, följ instruktionerna nedan:
1. Desinficera utsidan av injektionsflaskans gummipropp före insamling.
2. Fäst aseptiskt 5 mcm filternål (18G x 1½ ", 1,2 mm x 40 mm, medföljer) på en 1 ml spruta (medföljer). För in den trubbiga filternålen i mitten av locket tills den vidrör botten av injektionsflaskan.
3. Dra ut all vätska från injektionsflaskan genom att hålla den upprätt, något lutat för att underlätta fullständig uttagning.
4. Se till att sprutans kolv dras tillräckligt långt tillbaka när du tömmer injektionsflaskan för att helt tömma filternålen.
5. Lämna filternålen trubbig i injektionsflaskan och ta bort sprutan från den. Kassera filternålen efter att innehållet i injektionsflaskan har tagits ut och använd den inte för intravitreal injektion.
6. Fäst injektionsnålen (30G x ½ ", 0,3 mm x 13 mm, medföljer) säkert och aseptiskt på sprutan.
7. Ta försiktigt bort locket från injektionsnålen utan att koppla injektionsnålen från sprutan.
Obs! Håll injektionsnålens gula bas medan du tar av locket.
8. Släpp försiktigt ut luften från sprutan och justera dosen till 0,05 ml markerad på sprutan. Sprutan är klar för injektion.
Obs: Rengör inte injektionsnålen. Dra inte tillbaka kolven.
Efter injektionen ska du inte täcka nålen eller ta bort den från sprutan. Kassera den använda sprutan tillsammans med nålen i en lämplig behållare eller i enlighet med lokala krav.
07.0 INNEHAVARE AV GODKÄNNANDE FÖR FÖRSÄLJNING
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
Storbritannien
08.0 NUMMER FÖR FÖRSÄLJNINGSTILLSTÅND
EU/1/06/374/001
037608027
09.0 DATUM FÖR FÖRSTA GODKÄNNANDE ELLER FÖRNYELSE AV GODKÄNNANDET
Datum för det första godkännandet: 22 januari 2007
Datum för senaste förnyelse: 24 januari 2012
10.0 DATUM FÖR ÖVERSYN AV TEXTEN
05/2014


















.jpg)

